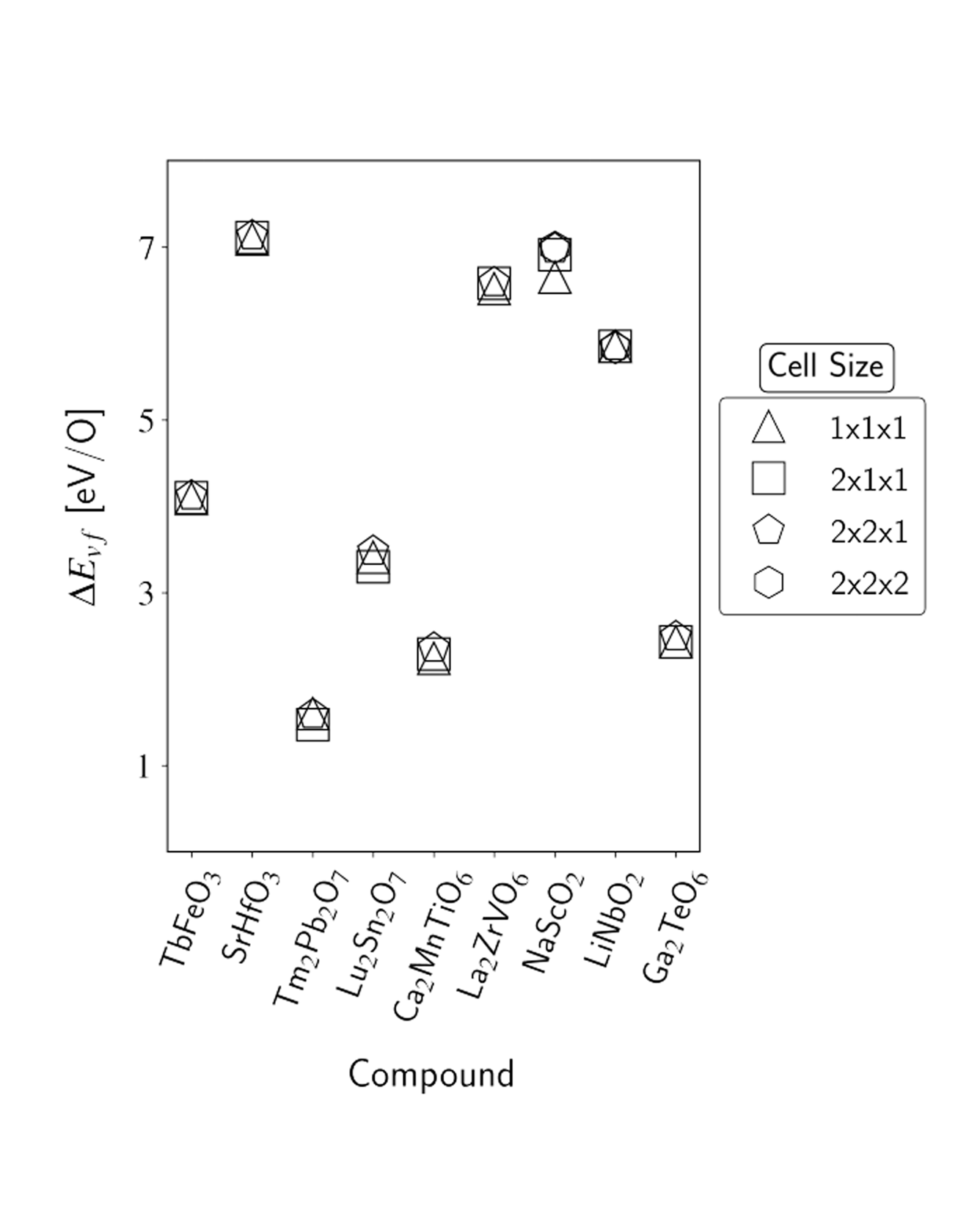
The oxygen vacancy formation energy (ΔEvf) governs defect concentrations alongside the entropy and is a useful metric to perform materials selection for a variety of applications. However, density functional theory (DFT) calculations of ΔEvf come at a greater computational cost than the typical bulk calculations available in materials databases due to the involvement of multiple vacancy-containing supercells. As a result, available repositories of direct calculations of ΔEvf remain relatively scarce, and the development of machine-learning models capable of delivering accurate predictions is of interest. In the present work, we address both such points. We first report the results of new high-throughput DFT calculations of oxygen vacancy formation energies of the different unique oxygen sites in over 1000 different oxide materials, with a large portion of the calculations, and of the discussion, focusing on perovskite-type and pyrochlore-type oxides. Together, the over 2500 ΔEvf calculations form the largest data set of directly computed oxygen vacancy formation energies to date, to our knowledge. We then utilize such a data set to train random forest models with different sets of features, examining both novel features introduced in this work and ones previously employed in the literature. We demonstrate the benefits of including features that contain information specific to the vacancy site and account for both cation identity and oxidation state and achieve a mean absolute error upon prediction of ∼0.3 eV/O, which is comparable to the accuracy observed upon comparison of DFT computations of oxygen vacancy formation energy and experimental results. Finally, we exemplify the predictive power of the developed models in the search for new compounds for solar-thermochemical water-splitting applications, finding over 250 new AA′BB′O6 double perovskite candidates. READ MORE