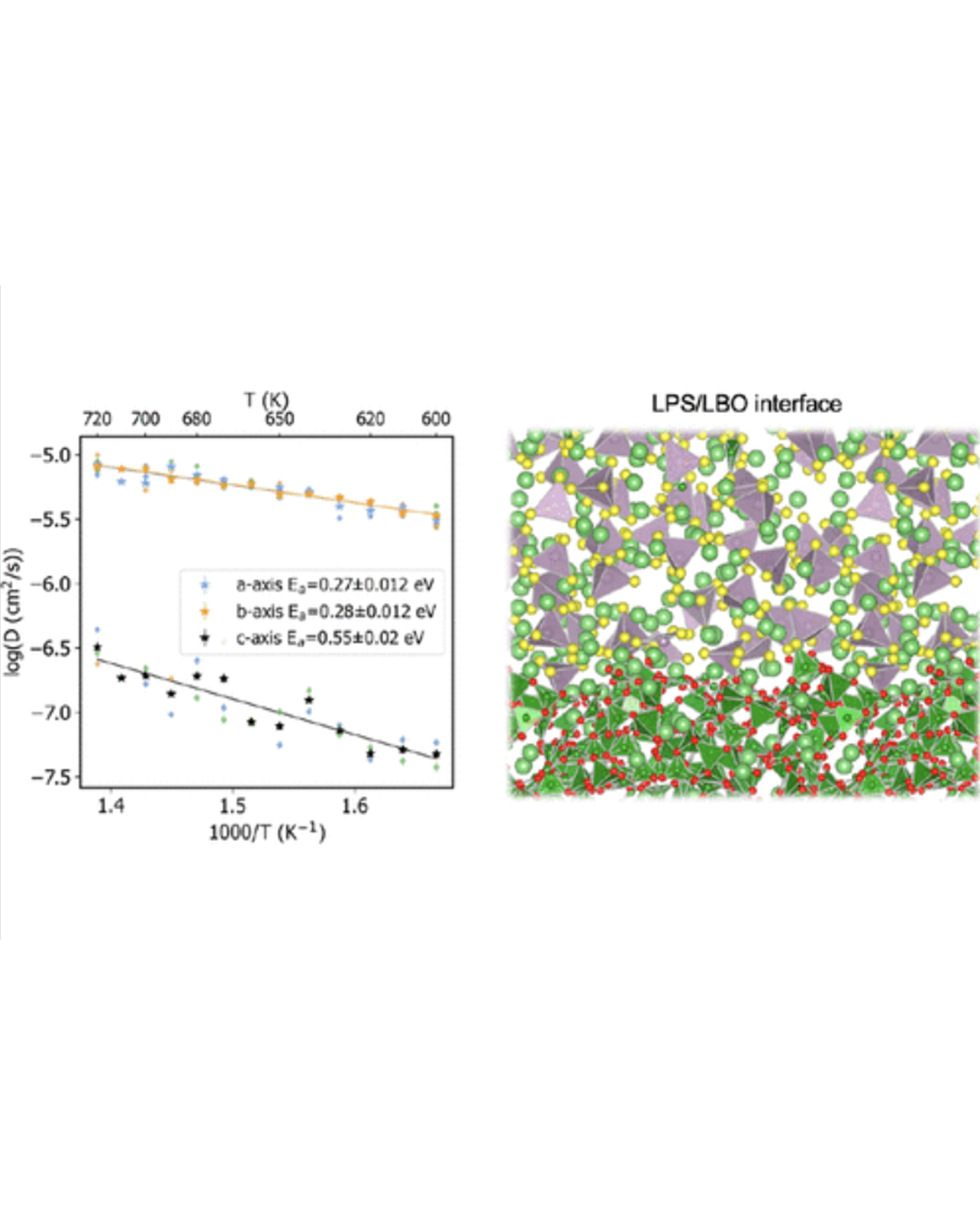
Non-crystalline solid materials have attracted growing attention in energy storage for their desirable properties such as ionic conductivity, stability, and processability. However, compared to bulk crystalline materials, fundamental understanding of these highly complex metastable systems is hindered by the scale limitations of density functional theory (DFT) calculations and resolution limitations of experimental methods. To fill the knowledge gap and guide the rational design of amorphous battery materials and interfaces, we present a molecular dynamics (MD) framework based on machine-learned interatomic potentials trained on the fly to study the amorphous solid electrolyte Li3PS4 and its protective coating, amorphous Li3B11O18. The use of machine-learned potentials allows us to simulate the materials at time and length scales that are not accessible to DFT while maintaining a near-DFT level of accuracy. This approach allows us to calculate amorphization energies, amorphous–amorphous interface energies, and the impact of the interface on lithium ion conductivity. This study demonstrates the promising role of actively learned interatomic potentials in extending the application of ab initio modeling to more complex and realistic systems such as amorphous materials and interfaces. READ MORE