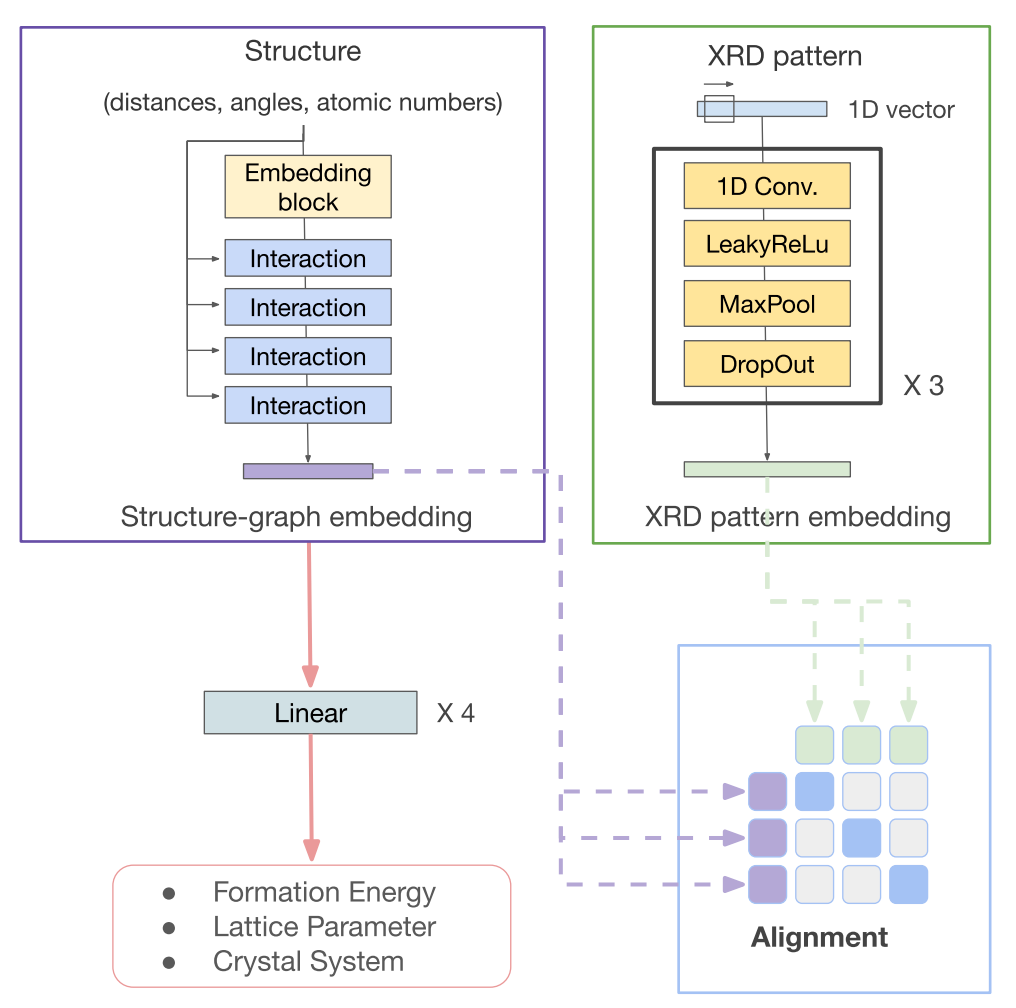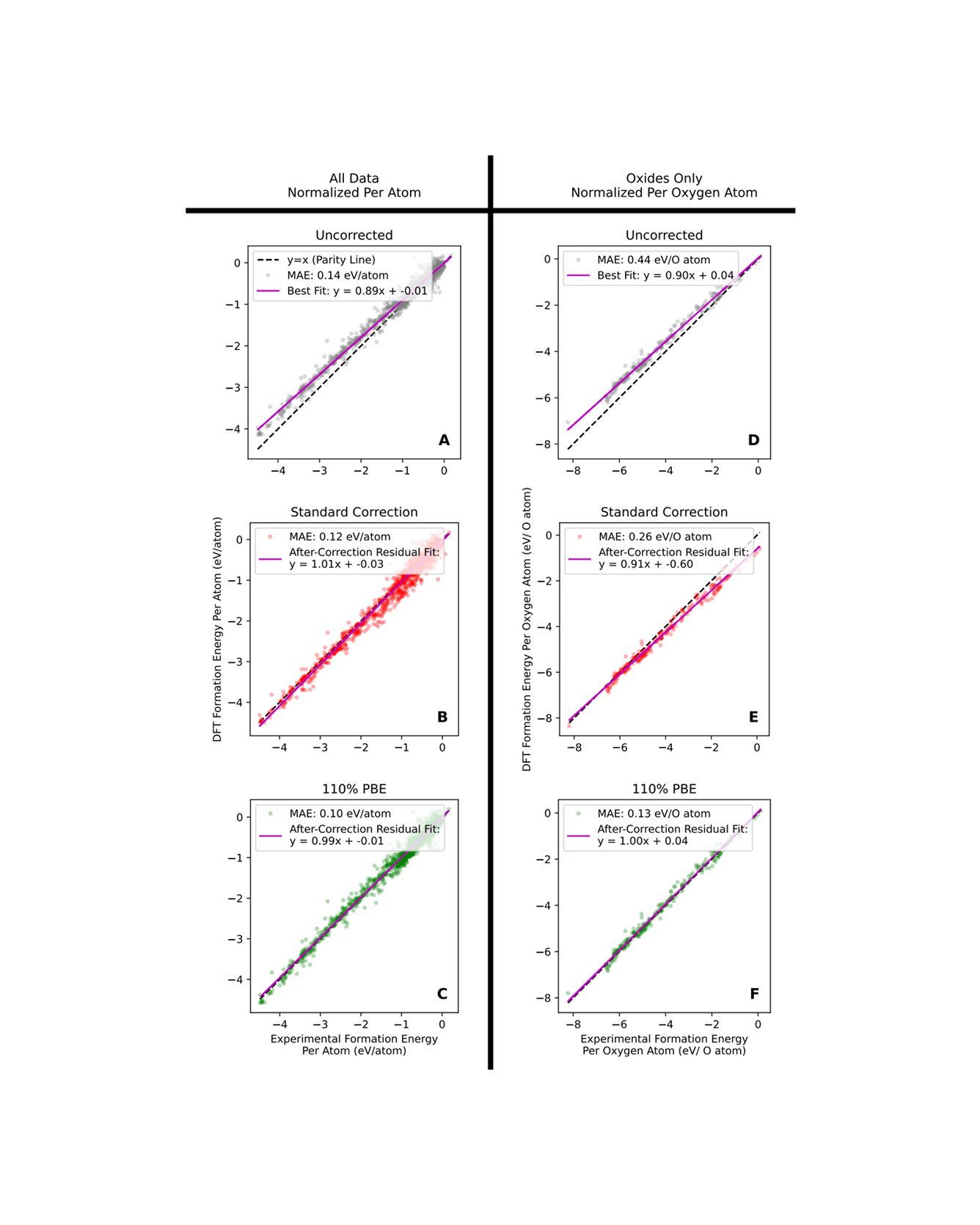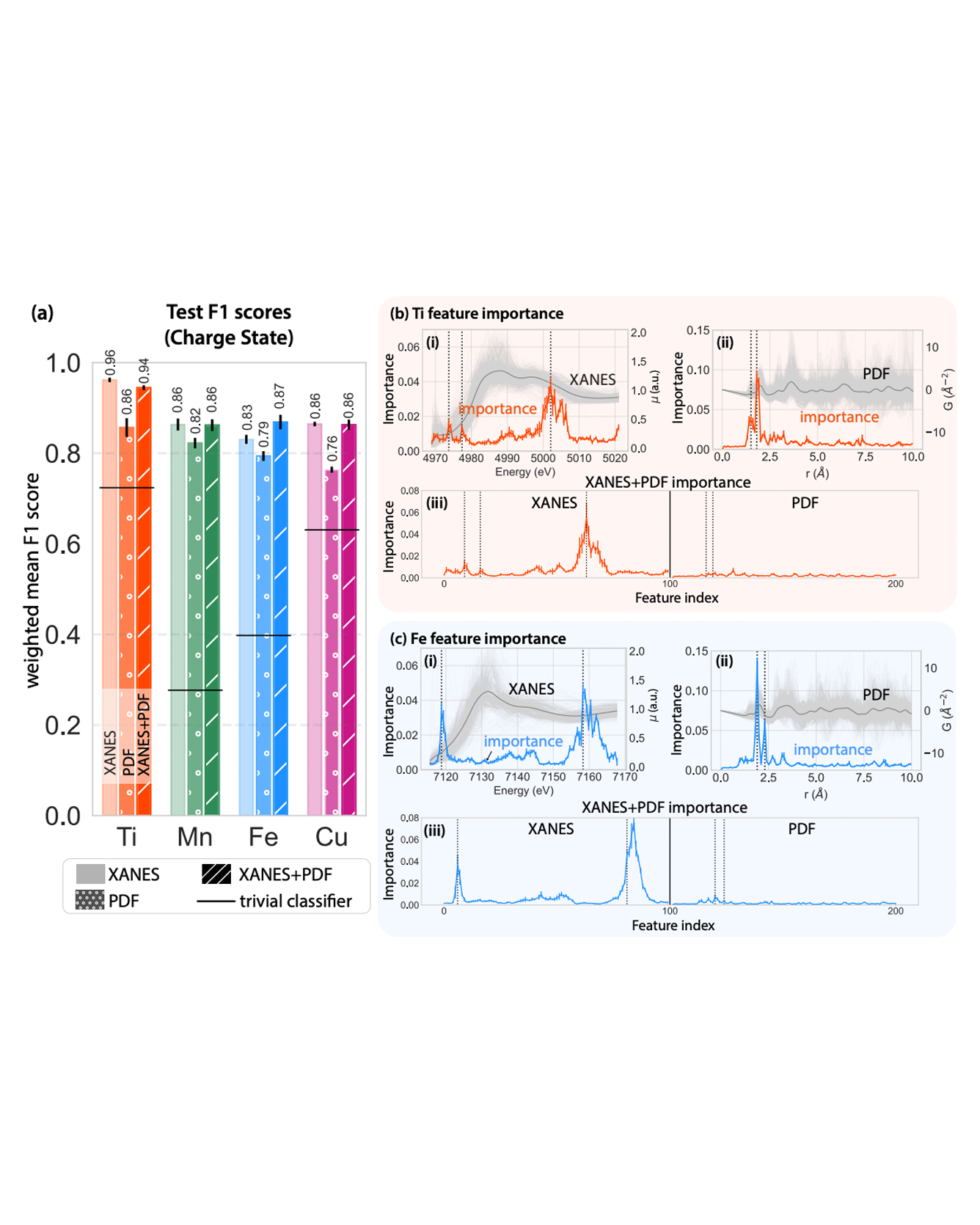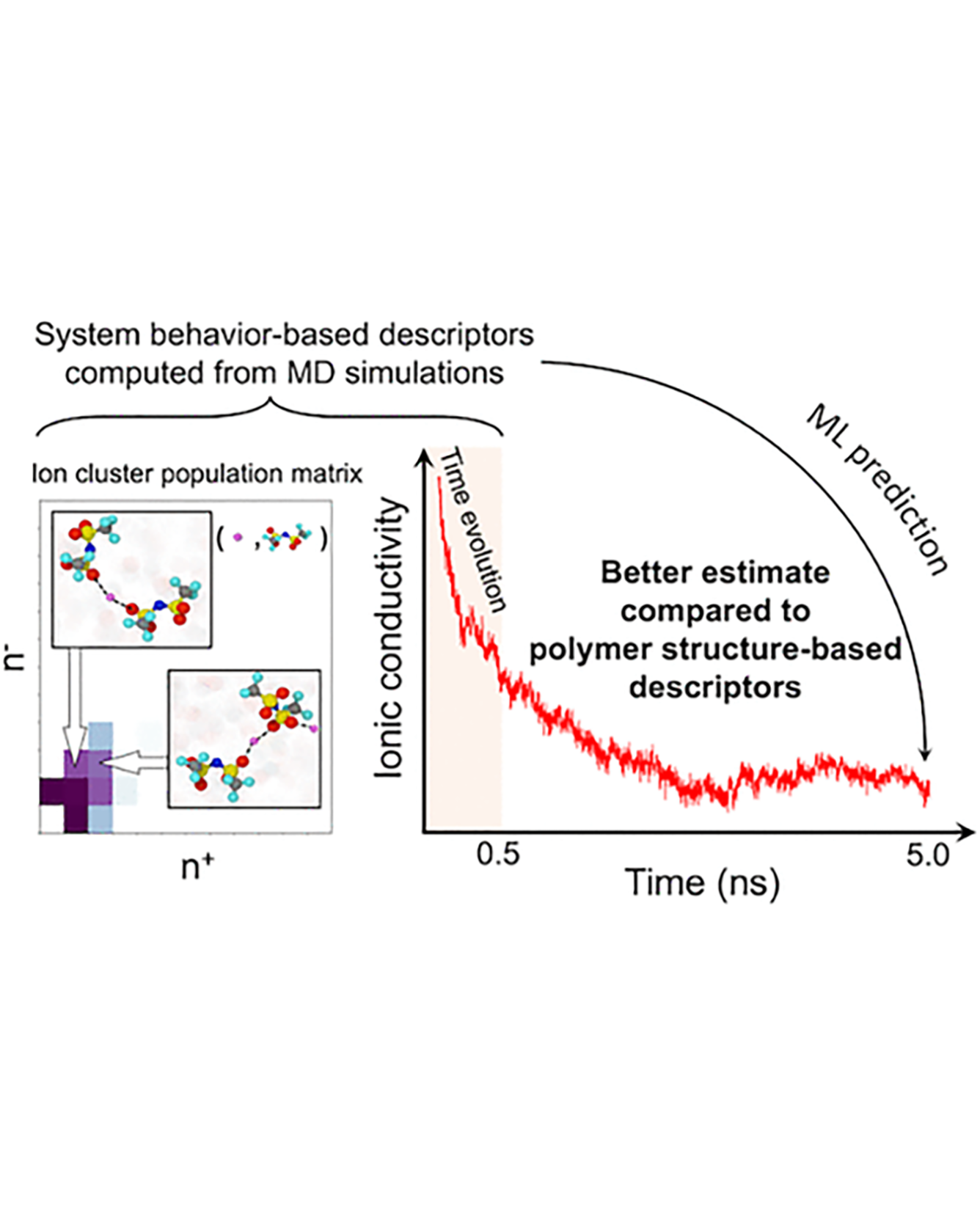
Molecular dynamics simulations are useful tools to screen solid polymer electrolytes with suitable properties applicable to Li-ion batteries. However, due to the vast design space of polymers, it is highly desirable to accelerate the screening by reducing the computational time of ion transport properties from simulations. In this study, we show that with a judicious choice of descriptors we can predict the equilibrium ion transport properties in LiTFSI–homopolymer systems within the first 0.5 ns of the production run of simulations. Specifically, we find that descriptors that include information about the behavior of the system, such as ion clustering and time evolution of ion transport properties, have several advantages over polymer structure-based descriptors, as they encode system (polymer and salt) behavior rather than just the class of polymers and can be computed at any time point during the simulations. These characteristics increase the applicability of our descriptors to a wide range of polymer systems (e.g., copolymers, blend of polymers, salt concentrations, and temperatures) and can be impactful in significantly shortening the discovery pipeline for solid polymer electrolytes. READ MORE